

Abstract
Introduction: Adenosine, an autacoid and metabolite of ATP, has been known to have anti-platelet properties. Of the 4 adenosine receptors (ARs), only A2A AR have been implicated in adenosines anti-platelet properties in human. A2A AR is a G-Protein Coupled Receptors associated with a stimulatory G-Protein (Gs) that can activate adenylyl cyclase (AC) and increase intracellular cAMP. An elevation of cAMP has been shown to inhibit platelet aggregation to natural stimuli. Regulation of intracellular cAMP is balanced between synthesis by adenylate cyclase and degradation by phosphdiesterases (PDE). There are 3 PDE subtypes found in platelets: PDE2, PDE3, and PDE5. However, it is not know which subtype(s) is (are) responsible for regulating cAMP level in human platelets after adenosine stimulation.
Materials and Methods: Platelet-rich plasma (PRP) was isolated from whole blood of human volunteers, and centrifuged at 200g for 10min. Light transmission aggregometry was performed after stimulation of platelets with 100uM ADP, with or without NECA (non-specific AR agonist), DPCPX (A1 AR antagonist), and Sch 58261 (A2A AR antagonist). PRP treated with NECA, DPCPX, Sch 58261, and PDE inhibitors (EHNA, E in figures, for PDE2, Trequinsin, T in figures, for PDE3, and 4-{[3'4'-(methylenedioxy) benzyl]amino}-6-methoxyqunazolin, 4 in figures, for PDE 5). Cyclic AMP was measured in platelets after treatment by liquid chromatography/ Tandem Mass Spectroscopy (Quantiva, ThrermoFisher) after treated with these drugs.
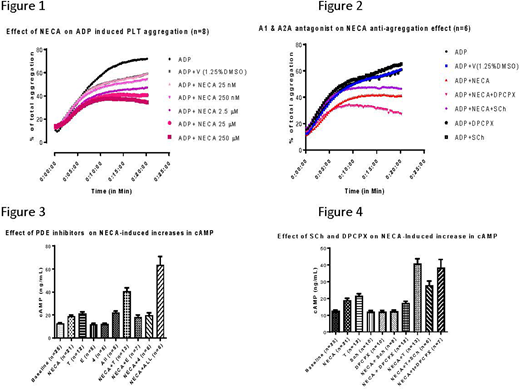
Results: ADP-induced platelet aggregation was inhibited in a dose dependent manner by the non-specific adenosine agonist, NECA (Figure 1) and the effect was blocked by A2A specific antagonist Sch 58261, not by the A1 AR antagonist, DPCPX (Figure 2). NECA inhibition of platelet aggregation was likely due to an elevation of intracellular cAMP (1 uM, 5min incubation, Figure 3). Inhibition of PDE3 alone, significantly increased intracellular cAMP, suggesting that basal PDE3 activity is present. PDE 3 inhibition combined with NECA elevated cAMP even higher than PDE inhibition or NECA alone (Figure 3), suggesting that NECA (A2A stimulation) effects PDE activity. Inhibition of PDE2 or 5 had no effect on basal or NECA stimulated cAMP (Figure 3). Inhibition of all 3 PDE (2,3,5) combined with NECA elevated cAMP to levels higher then NECA+ PDE3 inhibition, again suggesting that NECA maybe effecting the activity of the PDEs (Figure 3). The potentiation of cAMP by PDE3 inhibition + NECA was block by A2A, but not A1 antagonist (Figure 4) suggesting that the nonspecific adenosine agonist is elevating cAMP through A2A.
Conclusion: 1. In human platelets, NECA stimulates cAMP through A2A receptors and this elevation is likely due to an elevation in adenylate cyclase via Gs coupled to A2A. PDE3 is basally active and likely regulated by adenosine receptors.
No relevant conflicts of interest to declare.

